Катехоламин-зависимая полиморфная желудочковая тахикардия
Катехоламин-зависимая полиморфная желудочковая тахикардия
Введение
Катехоламин-зависимая полиморфная желудочковая тахикардия (Catecholaminergic ventricular tachycardia; CPVT) — наследственное заболевание, проявляющееся пароксизмами полиморфной или двунаправленной желудочковой тахикардии, возникающими на фоне физической нагрузки или эмоционального стресса (рис. 1), нередко протекающими с потерей сознания. Заболевание характеризуется злокачественным течением и высоким риском внезапной сердечной смерти при отсутствии адекватного лечения. Первые симптомы могут проявиться в возрасте от 2 до 36 лет (в среднем, в 8 лет). В 30–33% случаев в семейном анамнезе встречаются случаи внезапной сердечной смерти в младенчестве. У 60% больных развивается хотя бы один обморок в возрасте до 40 лет.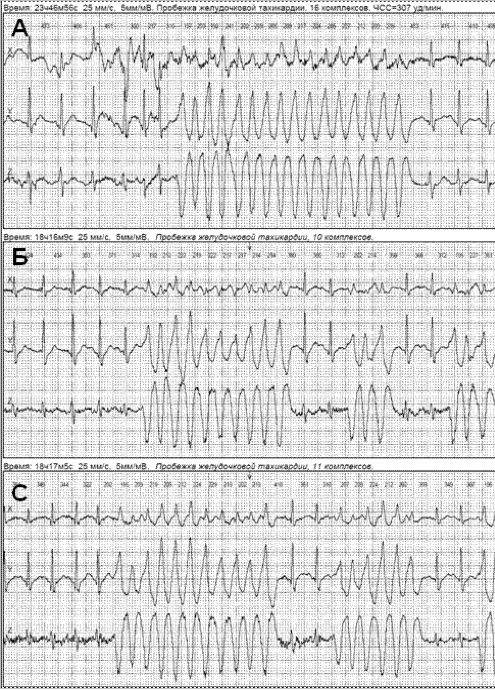
Рис. 1. Эпизоды катехоламин-зависимой полиморфной желудочковой тахикардии из 16 комплексов (А), 10 комплексов (Б), 11 комплексов (С), с частотой 260–307 уд./мин, зарегистрированные при суточном мониторировании ЭКГ по Холтеру во время эмоционального стресса (напряженный разговор по телефону) у больного 36 лет.
Эпидемиология
Истинная распространённость заболевания в общей популяции неизвестна, поскольку вне приступа у больных на ЭКГ отсутствуют какие-либо специфические изменения. По некоторым данным, она может достигать 1:10 000.Этиология
Катехоламин-зависимая полиморфная ЖТ является наследственным заболеванием, обусловленным нарушениями внутриклеточного обмена ионов Ca2+ вследствие мутаций генов RyR2 и CASQ2.У 30–35% больных мутации в генах RyR2 и CASQ2 отсутствуют, что указывает на генетическую гетерогенность заболевания и позволяет ожидать обнаружение новых мутаций, приводящих к возникновению заболевания, в дальнейшем. Описаны случаи обнаружения у больных катехоламин-зависимой полиморфной желудочковой тахикардией мутаций в генах KCNJ2, ANK2, TRDN и CALM1.Классификация
Описаны два генетических типа катехоламин-зависимой полиморфной ЖТ (табл. 1).- Первый тип заболевания обнаруживают у 65% пациентов. Болезнь наследуется по аутосомно-доминантному принципу и обусловлена мутациями гена райнодинового рецептора (Ryanodine receptor — RyR2). Этот рецептор является внутриклеточным кальциевым каналом, расположенным на мембране саркоплазматического ретикулума. Его дефект ведет к нарушению кальциевого гомеостаза, что сопровождается возникновением поздних постдеполяризаций, инициирующих развитие ЖТ.
- Второй тип катехоламин-зависимой полиморфной ЖТ обнаруживают у 3–5% больных. Заболевание наследуется по аутосомно-рецессивному принципу и обусловлено мутациями гена, кодирующего кальсеквестрин (calsequestrin — CASQ2) — белок, играющий роль основного кальциевого резервуара в саркоплазматическом ретикулуме кардиомиоцитов.
Таблица 1. Молекулярно-генетическая классификация катехоламин-зависимой желудочковой тахикардии

Диагностика
Проведение нагрузочной пробы на тредмиле или велоэргометре позволяет индуцировать пароксизм полиморфной желудочковой тахикардии у многих больных. При этом характерно появление и постепенное, по мере нарастания нагрузки, увеличение желудочковой эктопической активности вплоть до индукции устойчивого пароксизма желудочковой тахикардии или серии «пробежек» ЖТ. В некоторых случаях при невозможности проведения пробы с физической нагрузкой применяют фармакологические провокационные пробы с адреналином или изопротеренолом. Больным катехолоамин-зависимой полиморфной ЖТ характерна также индукция суправентрикулярных аритмий на фоне активации адренергических влияний.Важную роль в диагностике заболевания может играть длительное мониторирование ЭКГ с помощью переносных устройств и имплантируемых петлевых регистраторов.
Внутрисердечное ЭФИ при катехоламин-зависимой полиморфной ЖТ обычно неинформативно.
Молекулярно-генетические методы играют важную роль в диагностике заболеваний. Анализы на обнаружение мутаций в генах RyR2 и CASQ2 рекомендованы всем больным катехоламин-зависимой полиморфной ЖТ и пациентам, клинические проявления которых с большой вероятностью могут быть обусловлены этим заболеванием, особенно при отягощённом семейном анамнезе. В случае обнаружения у больного патогномоничной генетической мутации проведение скрининга, направленного на выявление этой мутации, рекомендовано всем близким родственникам, даже при отсутствии у них клинических проявлений заболевания.