Синдром Бругада
Синдром Бругада
Введение
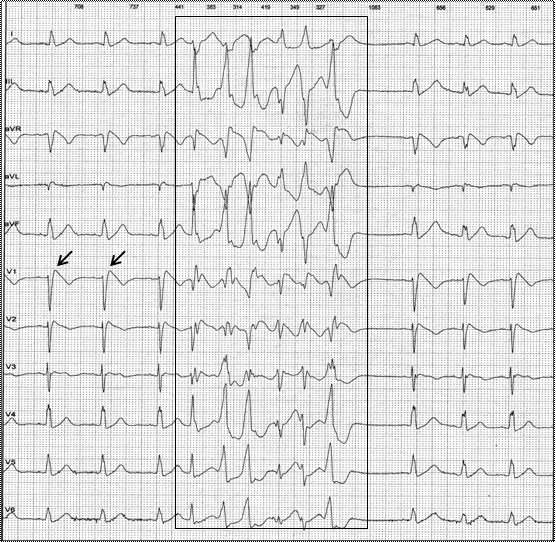
Синдром, характеризующийся ЭКГ признаками блокады правой ножки пучка Гиса с элевацией точки J и сегмента ST в правых прекордиальных отведениях и проявляющийся клинически рецидивирующими синкопальными состояниями, а также случаями внезапной сердечной смерти, которая наступает чаще у лиц мужского пола в возрасте 30–40 лет, описан P. Brugada и J. Brugada в 1992 г. Заболевание наследуется по аутосомно-доминантному типу, при этом характерна неполная пенетрантность генетических изменений.Желудочковые тахикардии, (преимущественно полиморфная, крайне редко — мономорфная) характеризующиеся высоким риском трансформации в фибрилляцию желудочков, являются основным клиническим проявлением синдрома Бругада. Характерно их возникновение в покое, во время ночного сна (рис. 1), что делает более вероятным их выявление с помощью ХМ ЭКГ, а не при стандартной записи ЭКГ. Одним из клинических проявлений, сопровождающих эти аритмические события, могут быть эпизоды хриплого (агонального) дыхания во сне. Желудочковые тахикардии могут провоцироваться лихорадочными состояниями, а также рядом лекарственных препаратов (см. табл. 1). Симптомы заболевания проявляются обычно у взрослых, а средний возраст возникновения случаев внезапной сердечной смерти составляет 41±15 лет. Кроме того, при синдроме Бругада чаще чем в общей популяции регистрируют случаи суправентрикулярных тахиаритмий.
Эпидемиология
Распространённость заболевания в общей популяции в настоящее время неизвестна. Существенно чаще оно встречается в странах Юго-Восточной Азии (Азиатско-Тихоокеанский регион), где его распространённость достигает 0,5–1:1000. Синдром Бругада (Brugada Syndrome, BrS) выявляется у лиц, не имеющих признаков органического заболевания сердца, у мужчин встречается в 8–10 раз чаще, чем у женщин, что, предположительно, обусловлено большей силой кратковременного выходящего тока ионов калия Ito (одного из токов, участвующих в формировании синдрома) и действием более высоких концентраций тестостерона.Этиология
Синдром Бругада вызывают генетические мутации, приводящие к уменьшению силы входящих натриевого (INa) и кальциевого (ICa,L) токов или увеличению силы выходящих калиевых токов (Ito,f, IKs, IK,ATP).Классификация
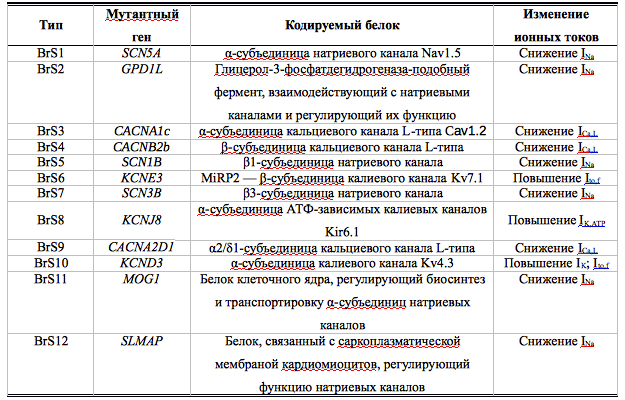
В настоящее время известны 12 генетических вариантов синдрома, они представлены в табл. 1. Молекулярно-генетические методы позволяют обнаружить мутации приблизительно у 1/3 больных с явными клинико-инструментальными проявлениями синдрома Бругада, что указывает на генетическую гетерогенность заболевания и позволяет предполагать открытие большого числа новых, не известных в настоящее время мутаций, в будущем. Наиболее распространены мутации гена SCN5A, которые обнаруживают почти у 30% пациентов.Таблица 1. Молекулярно-генетические типы синдрома Бругада
Диагностика
Основой диагностики синдрома Бругада является регистрация патогномоничных данному заболеванию изменений сегмента ST на ЭКГ при отсутствии структурного заболевания сердца и других состояний, при которых могут быть зарегистрированы подобные изменения ЭКГ (указаны далее). На основании характера изменений конечной части желудочкового комплекса выделяют три ЭКГ-типа феномена Бругада (табл. 2, рис 2).Таблица 2. ЭКГ-типы феномена Бругада

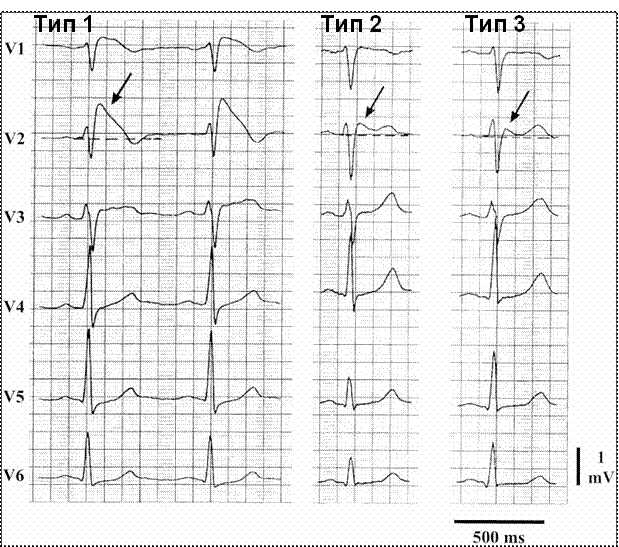
Регистрацию ЭКГ следует также проводить, располагая электроды правых прекордиальных отведений (V1–V2) выше стандартной позиции, вплоть до II межреберья. Выявление патогномоничных изменений ЭКГ в этих позициях имеет такую же диагностическую значимость, как и при стандартном расположении электродов. Изменения конечной части желудочкового комплекса, характерные для синдрома Бругада, могут иметь преходящий характер. Поэтому в тех случаях, когда имеющиеся записи ЭКГ не несут в себе признаков, в полной мере укладывающихся в диагностические критерии, но есть основание предполагать наличие синдрома Бругада, целесообразно проведение диагностических провокационных лекарственных проб с использованием блокаторов натриевых каналов, вводимых внутривенно, — аймалина (в дозе 1 мг/кг; в России не зарегистрирован) или прокаинамида (в дозе 10 мг/кг), позволяющих в части случаев «обнажить» признаки этого синдрома. Фармакологические провокационные пробы должны проводиться опытным медицинским персоналом при мониторировании ЭКГ больного и обязательной возможности организации реанимационных мероприятий в случае индукции опасных для жизни желудочковых аритмий под влиянием вводимых препаратов.
В соответствии с изменёнными диагностическими критериями, для постановки диагноза синдрома Бругада необходима регистрация на ЭКГ спонтанной или индуцированной лекарственными препаратами элевации сегмента ST по типу «свода» (тип 1) хотя бы в одном из правых прекордиальных отведений (V1–V2) при расположении электродов в типичном месте или установке их во II межреберье.
Методы молекулярно-генетической диагностики также имеют значение для диагностики заболевания, однако генетические мутации у больных синдромом Бругада удаётся обнаружить лишь приблизительно в 30% случаев, поэтому отрицательный результат генетического анализа не позволяет полностью исключить диагноз синдрома Бругада. В случае обнаружения у больного синдромом Бругада генетической мутации проведение скрининга, направленного на выявление этой мутации, рекомендовано всем близким родственникам, даже при отсутствии у них характерных этому заболеванию изменений ЭКГ. Проведение молекулярно-генетических исследований лицам, имеющим ЭКГ-изменения 2 и 3 типов, при отсутствии у них клинических проявлений синдрома Бругада и отягощённого по ВСС семейного анамнеза в настоящее время не рекомендовано.
Дифференциальная диагностика
Синдром Бругада следует дифференцировать от других возможных причин синкопальных состояний, учитывая относительно молодой возраст больных, прежде всего, от эпилепсии и вазо-вагальных обмороков, а также от других врождённых желудочковых нарушений ритма сердца.Необходимо также отметить, что схожие с синдромом Бругада изменения ЭКГ могут быть выявлены при целом ряде патологических состояний. Причинами «бругадоподобных» изменений ЭКГ могут быть:
- атипичная блокада правой ножки пучка Гиса;
- гипертрофия левого желудочка;
- феномен ранней реполяризации желудочков;
- острый коронарный синдром;
- аневризма левого желудочка;
- стенокардия Принцметала;
- острый перикардит;
- гемоперикард;
- тромбоэмболия лёгочной артерии;
- расслаивающая аневризма аорты;
- электролитные нарушения (гиперкалиемия, гиперкальциемия);
- гипотермия/гипертермия;
- аритмогенная дисплазия-кардиомиопатия правого желудочка;
- механическая компрессия выносящего тракта правого желудочка (например, опухоль органов средостения);
- передозировка трициклических антидепрессантов;
- кокаиновая интоксикация;
- различные заболевания центральной и вегетативной нервной системы (субарахноидальное кровоизлияние; геморрагический инсульт; атаксия Фридриха);
- мышечная дистрофия Дюшена–Беккера.